A new study links protein instability with the progression of a lethal degenerative disease: amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease. Using several biophysical techniques as well as small-angle x-ray scattering (SAXS) at the Advanced Light Source, researchers focused on the effects of mutations to a gene coding for a protein called superoxide dismutase (SOD). The study provides evidence that those proteins linked to more severe forms of the disease are less stable structurally and more prone to form clusters or aggregates, suggesting that strategies for stabilizing SOD proteins could be useful in treating or preventing SOD-linked ALS.
SOD1 mutations, the most studied factors in ALS, are found in about a quarter of hereditary ALS cases and seven percent of ordinary “sporadic” ALS cases. SOD-linked ALS has nearly 200 variants, each associated with a distinct SOD1 mutation. Scientists still don’t agree, though, on just how the dozens of different SOD1 mutations all lead to the same disease. One feature that SOD1-linked forms of ALS do have in common is the appearance of SOD clusters or aggregates in affected motor neurons and their support cells. Aggregates of SOD with other proteins are also found in affected cells, even in ALS cases that are not linked to SOD1 mutations.
In 2003, the researchers had proposed the “framework destabilization” hypothesis. In this view, ALS-linked mutant SOD1 genes all code for structurally unstable forms of the SOD protein. Inevitably some of these unstable SOD proteins lose their normal folding enough to expose sticky elements that are normally kept hidden, and they begin to aggregate with one another, faster than neuronal cleanup systems can keep up—and that accumulating SOD aggregation somehow triggers disease. In this study, the researchers used advanced biophysical methods to probe how different SOD1 gene mutations in a particular genetic ALS “hotspot” affect SOD protein stability.
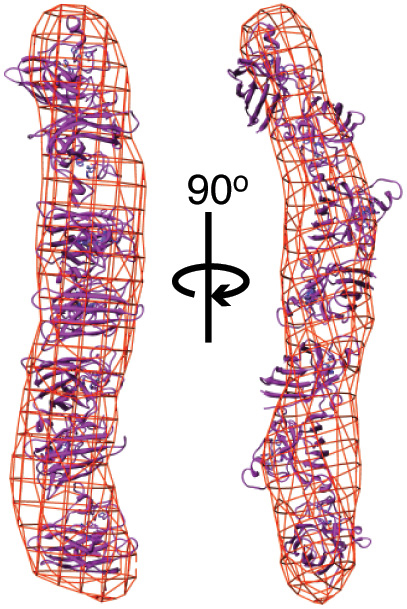
To start, they examined how the aggregation dynamics of the best-studied mutant form of SOD, known as SOD G93A, differed from that of non-mutant, “wild-type” SOD. To do this, they developed a method for gradually inducing SOD aggregation, which was measured using small-angle x-ray scattering (SAXS) at Beamline 12.3.1 of the Advanced Light Source. The G93A SOD aggregated more quickly than wild-type SOD, but more slowly than an SOD mutant called A4V that is associated with a more rapidly progressing form of ALS. Subsequent experiments with G93A and five other G93 mutants revealed that the mutants formed long, rod-shaped aggregates, compared to the compact folded structure of wild-type SOD. The mutant SOD proteins that more quickly formed longer aggregates were again those that corresponded to more rapidly progressing forms of ALS.
What could explain these SOD mutants’ diminished stability? Further tests focused on the role of a copper ion that is normally incorporated within the SOD structure and helps stabilize the protein. Using two other techniques—electron-spin resonance spectroscopy and inductively coupled plasma mass spectrometry—the researchers found that the G93-mutant SODs had a reduced ability to retain copper under mildly stressing conditions. The copper deficiency in these mutants in turn promotes aggregation in a manner strikingly consistent with their correlated clinical severities.
Along with others in the field, the researchers suspect that deviant interactions of mutant SOD trigger inflammation and disrupt ordinary protein trafficking and disposal systems, stressing and ultimately killing affected neurons. The researchers also plan to confirm the relationship between structural stability and ALS severity in other SOD mutants. If their hypothesis is correct, future therapies to treat SOD-linked ALS need not be tailored to each individual mutation—they should be applicable to all of them.
Contacts: Elizabeth Getzoff and John Tainer
Research conducted by: A.J. Pratt, D.S. Shin, and J.A. Tainer (The Scripps Research Institute and Berkeley Lab); G.E. Merz, P.P. Borbat, J.H. Freed, and B.R. Crane (Cornell University); R.P. Rambo and K.N. Dyer (Berkeley Lab); W.A. Lancaster, F.L. Poole II, and M.W.W. Adams, (University of Georgia); and E.D. Getzoff (The Scripps Research Institute).
Research funding: National Institutes of Health, National Science Foundation, Skaggs Institute for Chemical Biology, Buck Institute for Research on Aging, and the U.S. Department of Energy (DOE), Office of Biological and Environmental Research. Operation of the ALS is supported by DOE, Office of Basic Energy Sciences.
Publications about this research: A.J. Pratt, D.S. Shin, G.E. Merz, R.P. Rambo, W.A. Lancaster, K.N. Dyer, P.P. Borbat, F.L. Poole II, M.W.W. Adams, J.H. Freed, B.R. Crane, J.A. Tainer, and E.D. Getzoff, “Aggregation propensities of superoxide dismutase G93 hotspot mutants mirror ALS clinical phenotypes,” Proc. Natl. Acad. Sci. U.S.A. 111, E4568 (2014); and G.E. Merz, P.P. Borbat, A.J. Pratt, E.D. Getzoff, J.H. Freed, and B.R. Crane, “Copper-based pulsed dipolar ESR spectroscopy as a probe of protein conformation linked to disease states,” Biophys. J. 107, 1669 (2014).
ALS SCIENCE HIGHLIGHT #309
![]()
![]()