The cancer drug Gleevec is extremely specific, binding and inhibiting only the cancer-causing tyrosine protein kinase Blc-Abl, while not targeting homologous protein kinases found in normal, healthy cells. It has been widely used to fight colon cancers and chronic myeloid leukemia. The protein kinase Abl is involved in regulating cell growth. Protein kinases have in general been the target of many cancer drug designs, since they are responsible for so many cellular signaling functions. However Gleevec is one of the very few success stories of a specific kinase inhibitor. The proteins look nearly identical, and particular the Gleevec binding pockets between the tight binding Abl kinase and a close homologue Src kinase that in contrast binds Gleevec extremely weak are nearly identical, which begs the question: why exactly does Gleevec bind so well to Abl and not Src?
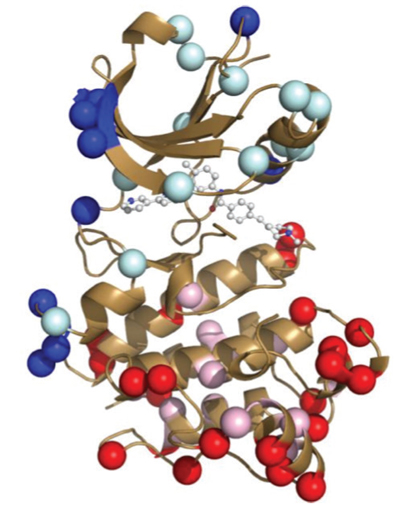
The actual structures of Abl and Src, both alone and bound to Gleevec, have been known for some time. Indeed, many structural studies on these proteins with designed mutations have also previously sought to understand why exactly Gleevec binds preferentially to Abl but, until now, this mechanism was not understood. As it turns out, structure alone was not enough to solve the puzzle.
In order to study the amazing specificity of Gleevec, researchers working at the ALS first turned to something that at first glance appears completely unrelated: the evolutionary history of the two proteins Abl and Src. Using 76 modern tyrosine kinase structures in a statistical method called Bayesian phylogenetic analysis, they reconstructed the “family tree” for the protein. The common ancestor of Abl and Src, a protein they called ANC-AS, existed about a billion years ago. Since that time, the protein accumulated many mutations in the evolution of many different organisms, diverging into the two modern proteins Abl and Src, which are different by 146 amino acids, though retaining the same overall physical structure. The group then reconstructed four protein kinases along the evolutionary tree, including the common ancestor, and grew those proteins in the lab.
Measuring the kinetics of Gleevec binding to each of these proteins gave the second important clue. It turns out that binding involves two steps: the initial binding, and then a slow “induced-fit” process in which the protein changes conformation until the drug fully binds. We tend to think of proteins as being in a static frozen state, and indeed this is how protein crystallography structures depict them. But in reality, proteins are in a constant state of motion, hence are constantly interconverting between structures! This dynamic nature is essential for biological function. Those rearrangements can be subtle: one small loop of a protein “flopping around,” for instance, or they can be as large as entire sections of a protein unfolding and then folding back again. How much time a protein spends in one structural state relative to another determines its conformation equilibrium. Abl, it turns out, is just different enough from Src that Gleevec can induce a large shift in the equilibrium between two drug-bound structures. This induced-fit step of the process was what gradually changed throughout evolution to make Gleevec much more selective to Abl than to Src.
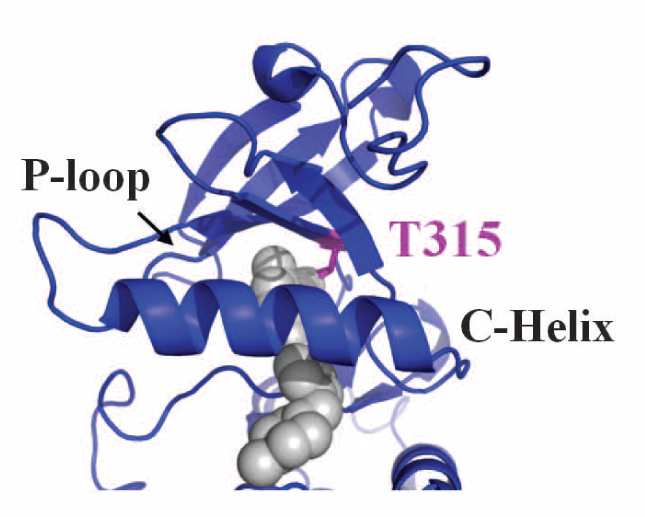
Finally, crystallography was used to determine the actual physical structure of the ancestral tyrosine kinase protein ANC-AS bound to Gleevec, and to compare it to the structures of the modern-day kinase bound to Gleevec. The structural analysis showed an extensive hydrogen-bonding network that is present in the ancestral protein and in Src, but is not present in Abl. The authors hypothesize that the lack of this hydrogen-bonding network is what allows a specific loop in the Abl protein to be more flexible, which in turn allows Gleevec to better induce protein conformational changes in Abl vs Src.
Gleevec is an exceptionally selective drug for certain cancers, targeting the Abl tyrosine kinase in cancer cells. Of course, Gleevec did not drive evolution of this tyrosine kinase. Gleevec was designed to fit into the binding pocket of the protein, and serendipitously bound preferentially to Abl over Src. But uncovering the reasons for this points to new possibilities for drug design. Perhaps drugs can be designed that will target a specific subset of structures within a conformational equilibrium, rather than designing drugs that fit well to just one model. Perhaps investigating the evolutionary family tree for a family of proteins will shed light into the evolution of differences in the energy landscapes of modern protein kinases that can then be exploited by cancer drugs with high specificity. This study opens the door for new types of studies like this in the on-going search for cancer therapies.
Contact: Dorothee Kern
Research conducted by: C. Wilson, R. V. Agafonov, M. Hoemberger, S. Kutter, A. Zorba, J. Halpin, V. Buosi, R. Otten, D. Waterman, and D. Kern (Howard Hughes Medical Institute, Brandeis University); D. L. Theobald (Brandeis University).
Research funding: Howard Hughes Medical Institute, U.S. Department of Energy (DOE), Basic Energy Sciences (BES), Catalysis Science Program. Operation of the ALS is supported by the DOE BES.
Publication about this research: C. Wilson, R. V. Agafonov, M. Hoemberger, S. Kutter, A. Zorba, J. Halpin, V. Buosi, R. Otten, D. Waterman, D. L. Theobald, D. Kern, “Using ancient protein kinases to unravel a modern cancer drug’s mechanism,” Science 347, 882 (2015).
ALS SCIENCE HIGHLIGHT #314
![]()
![]()