A major challenge of computational structural biology has been to create, from scratch, new proteins with heretofore unobserved three-dimensional structures. A collaboration from the University of Washington, Seattle, the University of North Carolina, Chapel Hill, and the Fred Hutchinson Cancer Research Center has now developed and demonstrated a methodology for protein-structure prediction and design by creating the first artificial globular protein with a novel topology, a 93-residue protein called Top7. Significantly, the x-ray structure of Top7 agreed almost precisely with the structure specified by the computational model.

Protein function depends on the complex geometries assumed as sequences of amino acids (each comprising a carboxyl group, an amino group, and a side chain) link into chains of residues that form local structural units such as α helices and β strands, and finally fold into compact, three-dimensional, globular domains and multidomain structures. Previous computational protein design attempts have focused primarily on redesigning the sequence of naturally occurring proteins to enhance their stability or to achieve new functionality.
These methods generally start with a known, high-resolution structure of the target protein and then try to optimize the packing of different amino-acid side chains while keeping the backbone template (carboxyl and amino groups) fixed to arrive at new low-energy sequence solutions. The key features of these methods are an efficient search protocol for sampling the theoretically vast number of sequence permutations and an energy function designed to model the physical forces that hold natural proteins together.
The collaborators extended these concepts in their RosettaDesign method. However, they were faced with the additional challenge of sampling protein-backbone structural space as well as sequence space, since their goal was the creation of a novel fold (where no natural backbone template was available). To this end, they constructed RosettaDesign to iterate between full-scale optimization of the sequence for a fixed-backbone conformation and gradient-based optimization of the backbone coordinates for a fixed sequence. Beginning with a simple back-of-the-envelope sketch of the target, a novel α/β fold, and this protocol, they designed Top7, a 93-residue α/β protein with a topology not observed in the Protein Structure Database (PDB), i.e., an artificial protein.
By means of a variety of biophysical techniques, the researchers determined the synthesized Top7 protein to be monomeric, highly soluble, and extremely stable to chemical and thermal denaturation. Preliminary NMR analysis also showed that Top7 had a rigid structure consistent with the target topology. Finally, thanks to the ALS Howard Hughes Medical Institute Beamline 8.2.1, they solved an x-ray structure of a single selenomethionyl-substituted variant of Top7 to 2.5-Å resolution with single-wavelength anomalous diffraction (SAD) data.
This high-resolution crystal structure revealed that the Top7 protein adopted the designed topology and in fact was strikingly similar to the design model at atomic resolution (1.17-Å root-mean-square deviation or RMSD over all backbone atoms). The two models differ most in the region surrounding the first N-terminal (amino-group end) hairpin, but even here the all-atom RMSD did not exceed 2.8 Å. In contrast, the C-terminal (carboxyl-group end) halves of the crystal structure and the designed model are very similar, and core side-chain atoms are virtually superimposable.
The successful design of Top7 has two major implications. First, it is a strong validation of the understanding and description of the energetics of proteins and other macromolecules, much of which, incidentally, has been a consequence of the determination of high-resolution structures of those macromolecules. Second, it suggests that the development of protein therapeutics and molecular machines need not be limited to the structures sampled by the biological evolutionary process.
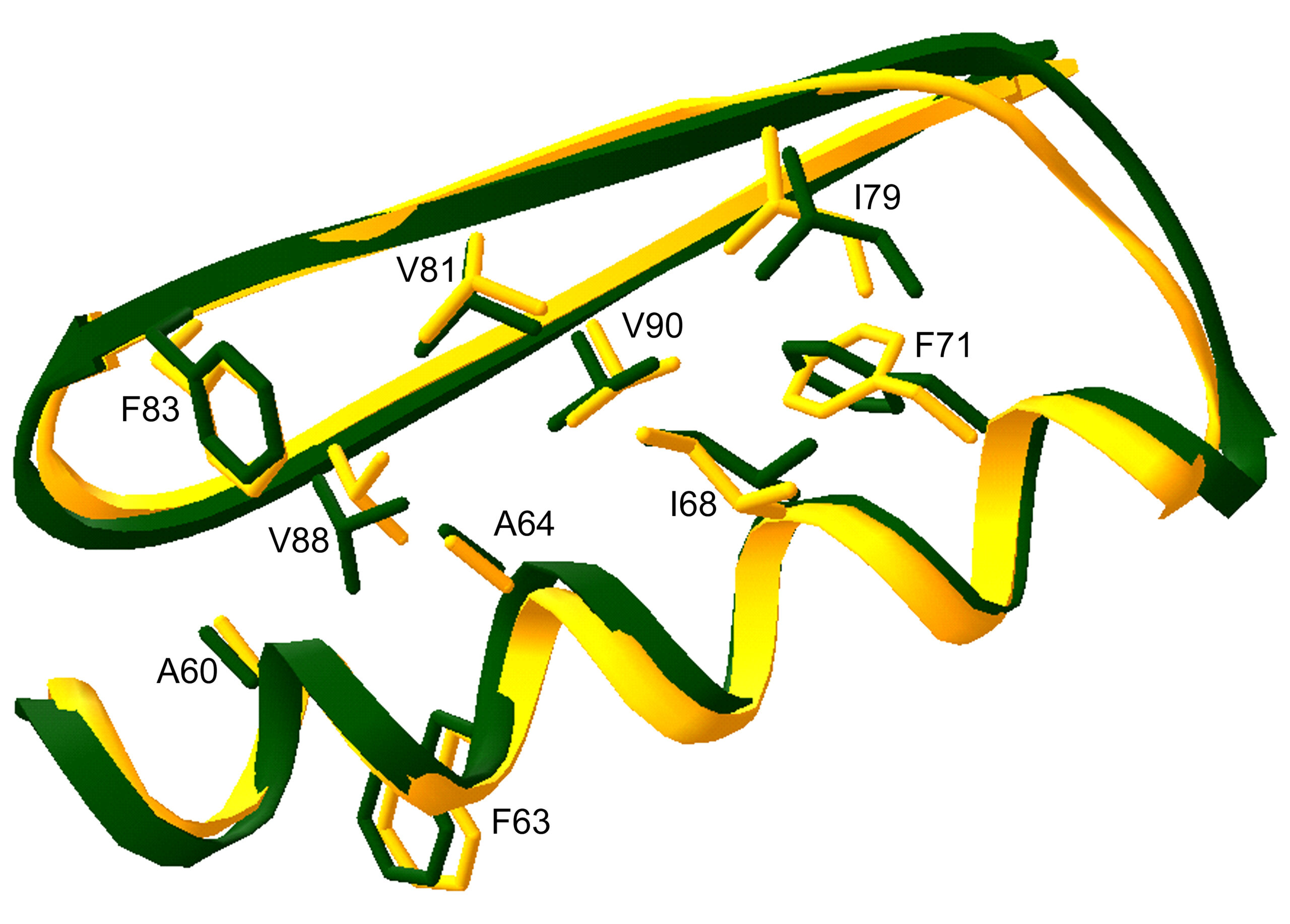
Contact: David Baker
Research conducted by G. Dantas, G. Varani, and D. Baker (University of Washington, Seattle); B. Kuhlman (University of North Carolina, Chapel Hill); and G.C. Ireton and B.L. Stoddard (Fred Hutchison Cancer Research Center, Seattle).
Research funding: The National Institutes of Health and the Cancer Research Fund of the Damon Runyun–Walter Winchell Foundation. Operation of the ALS is supported by the U.S. Department of Energy, Office of Basic Energy Sciences.
Publication about this research: B. Kuhlman, G. Dantas, G.C. Ireton, G. Varani, B.L. Stoddard, and David Baker, “Design of a novel globular protein fold with atomic-level accuracy,” Science 302, 1364 (2003), doi:10.1126/science.1089427.
ALS SCIENCE HIGHLIGHT #76
![]()
![]()