Naturally occurring proteins—chains of amino acids that fold into functional, three-dimensional shapes—are believed to represent just a small fraction of the universe of all possible permutations of amino-acid sequences and folds. How can we begin to systematically sift through those permutations to find and engineer from scratch (de novo) proteins with the characteristics desired for medical, environmental, and industrial purposes? To address this question, a team led by researchers from the Institute for Protein Design at the University of Washington have published a landmark study that used both protein crystallography and small-angle x-ray scattering (SAXS) at the ALS to validate the computationally designed structures of novel proteins with repeated motifs. The results show that the protein-folding universe is far larger than realized, opening up a wide array of new possibilities for biomolecular engineering.
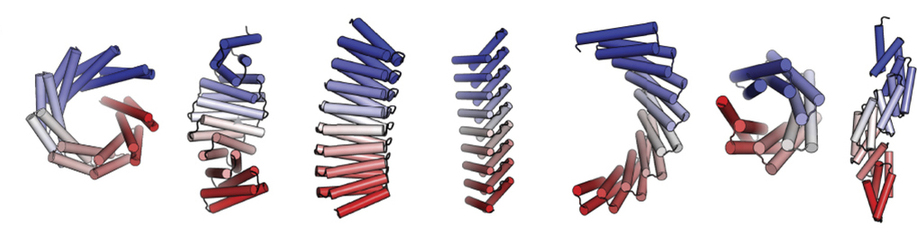
Repeat proteins composed of multiple copies of a modular unit are widespread in nature and play critical roles in molecular recognition, signaling, and other essential biological processes. The shape and curvature of a repeat protein’s overall structure is defined by interactions between adjacent units, and customization of these in existing protein families has allowed for the control of curvature and the design of new architectures. These naturally occurring families may cover all stable repeat-protein structures that can be built from the 20 amino acids or, alternatively, natural evolution may only have sampled a subset of what is possible.
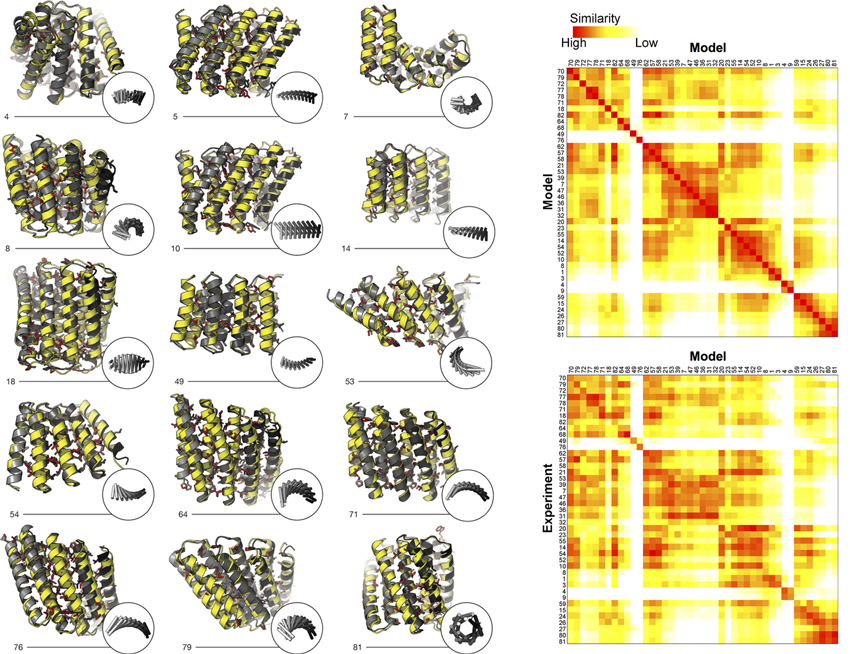
In this work, the researchers used computational protein design to investigate the space of folded structures that can be generated by the modular assembly of helices and loops (linkers). Combinations of these, varying helix and linker lengths, were systematically sampled and extended into over a hundred designs using computer modeling and analysis software. Eighty-three of the designs with very low correlation to known existing structures were selected for experimental characterization. Of the 83 designs, 74 were expressed in a soluble form and showed helical content. Fifteen of the proteins were successfully crystallized and their structures were solved at Beamlines 8.2.1 (Berkeley Center for Structural Biology) and 8.3.1 (University of California). These repeat proteins are among the largest crystallographically validated protein structures designed completely de novo, ranging in size from 171 to 238 residues. The crystal structures illustrate the wide range of twists and curvatures sampled by the repeat-protein generation process and the accuracy with which these proteins can be designed.
To characterize the structures of proteins that were resistant to crystallization, the researchers used SAXS at Beamline 12.3.1 (the Structurally Integrated Biology for Life Sciences, or “SIBYLS,” beamline). While this technique cannot specify atomic structure, it can provide information about tertiary (overall shape) and quaternary (subunit coupling) features. SAXS experiments can quickly screen protein structures in the solution state, distinguishing conformational states and characterizing even flexible macromolecules. Because the protein solutions are dilute and the molecules scatter weakly, the high brightness of the ALS is crucial to the process of data gathering and analysis. Structural similarity maps (SSMs) were generated to facilitate the identification of similarities and differences at a glance. For 43 of the designs, the radius of gyration, molecular weight, and distance distributions computed from the SAXS data matched those computed from models.
The crystallographic and SAXS data together structurally validated more than half of the 83 designs that were experimentally characterized, showing that a wide range of novel repeat proteins can be generated by tandem repeating a simple helix–loop–helix–loop building block. The work achieved key milestones in computational protein design: the design protocol is completely automatic, the folds are unlike those in nature, more than half of the experimentally tested designs have the correct overall structure as assessed by SAXS, and the crystal structures demonstrate precise control over backbone conformation for proteins of over 200 amino acids.
Contacts: David Baker, Greg Hura, and Susan Tsutakawa
Research conducted by: T.J. Brunette, F. Parmeggiani, and P.-S. Huang (University of Washington); G. Bhabha and D.C. Ekiert (UC San Francisco); S.E. Tsutakawa (Berkeley Lab); G.L. Hura (Berkeley Lab and UC Santa Cruz); J.A. Tainer (Berkeley Lab and University of Texas); D. Baker (University of Washington and Howard Hughes Medical Institute).
Research funding: National Science Foundation, Defense Threat Reduction Agency, Air Force Office of Scientific Research, and Howard Hughes Medical Institute. Operation of the ALS is supported by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences (BES).
Publication about this research: T.J. Brunette, F. Parmeggiani, P.-S. Huang, G. Bhabha, D.C. Ekiert, S.E. Tsutakawa, G.L. Hura, J.A. Tainer, D. Baker, “Exploring the repeat protein universe through computational protein design,” Nature 528, 580 (2015).
ALS SCIENCE HIGHLIGHT #328
![]()
![]()