Curved β sheets are basic building blocks of many protein cavities that, by serving as binding sites for other molecules, are essential to protein function. After analyzing classic protein formations and running folding simulations, researchers designed a series of novel proteins with curved β sheets, inspired by naturally occurring protein superfamilies. They then compared the predicted models to physical examples of these designed proteins using x-ray crystallography. All of the structures closely matched the predicted models, showing that β-sheet curvature can be controlled with atomic-level accuracy. The discovery opens the door to the design of proteins capable of entirely new functions, from improved diagnostic tests and medical treatments to more-efficient catalysis of chemical reactions for industrial processes.
In designing a new ligand-binding protein or enzyme acting on a particular molecule, researchers have traditionally tried to repurpose natural proteins. They look for proteins with cavities having roughly the desired geometry and incorporate mutations to achieve the function of interest. However, this strategy is known to have several limitations: the ideal geometry may not be found and/or the incorporated mutations may change the cavity geometry or be detrimental to the protein’s stability. To overcome these limitations, it would be more efficient to design a protein from scratch, with a geometry customized to the target of interest.
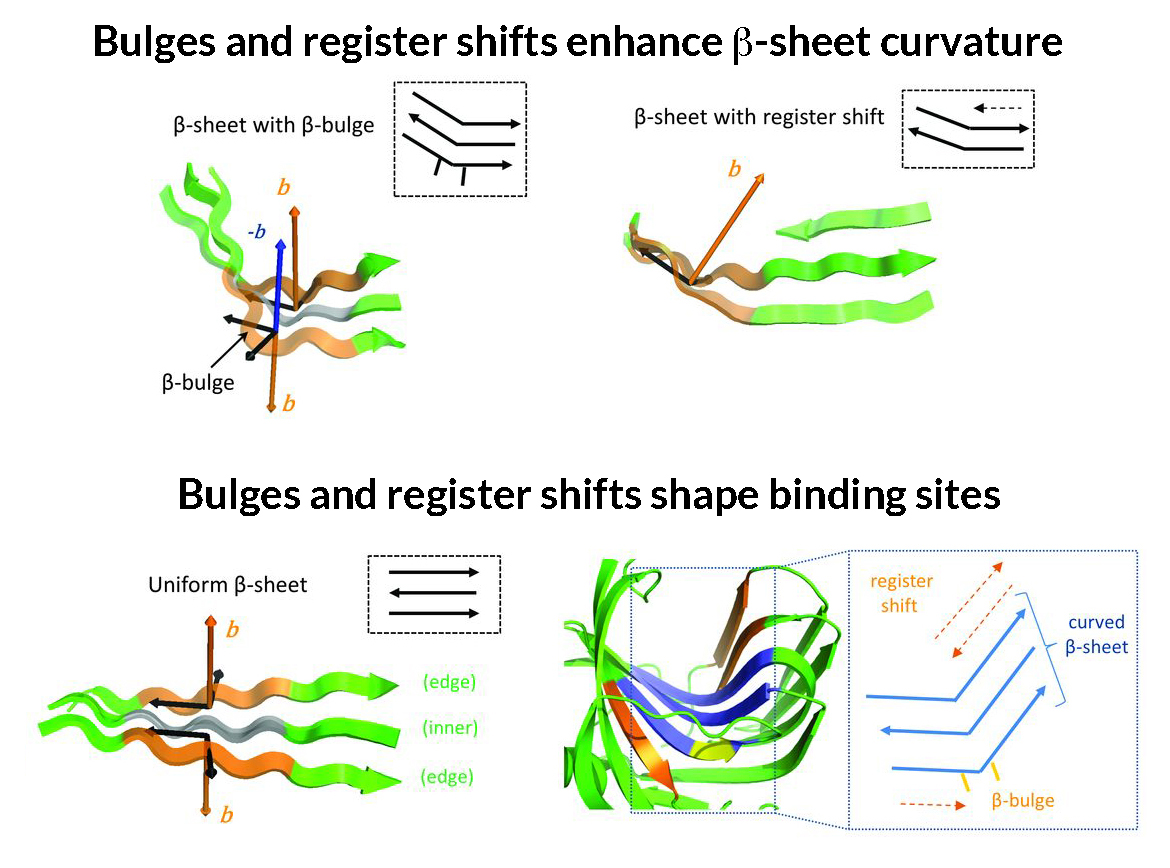
Previously, the computational design of proteins from scratch had been limited to structures that were too compact to accommodate cavities. In this work, researchers deciphered the key rules that determine the shape of structures formed by β sheets in natural proteins. In particular, they described the rules that govern how these β sheets bend and wrap to form the cavities of various sizes and shapes that play a key role in protein–protein or protein–molecule interactions. For example, β sheets (normally flat) will bulge if hydrophobic (or -philic) residues are placed next to each other in the protein chain. Also, by terminating the bonding between adjacent strands (a “register shift”), a bend can be created. The researchers then incorporated these rules into existing computational protein design methods and built new types of protein structures having the shapes they were targeting.
To verify that the resulting proteins had the desired properties and adopted the modeled structure, the researchers experimentally characterized them with a variety of techniques. The atomic structures were verified using high-resolution x-ray crystallography at Beamline X4C of the National Synchrotron Light Source and at ALS Beamline 8.2.1. Of the nine solved structures described in this work, six were obtained at the ALS. High-resolution structural analysis is essential for evaluating the accuracy of computational designs and quickly becomes the bottleneck in protein design studies. The ability to quickly screen many crystals at the ALS enhanced the probability of solving the structures.
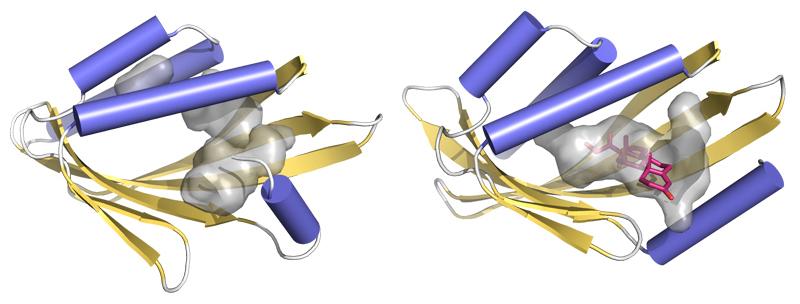
Interpretation and analysis of the crystallographic data showed that the experimental protein structures were very close to the computational models, validating the new design approach. The researchers were surprised to find that a few small molecules from the crystallization solution had found their way into the cavities of one of the designed proteins—a good indication of the feasibility of follow-up plans for designing an active site there with little change from the initial structure. Now that they have shown that they can design these types of proteins, the researchers plan to use this method to create novel enzymes that could result in reduced chemical waste and more-efficient chemistry in general, as well as sensors for the detection of food and environmental toxins. Overall, the new strategy allows designers to depend less on natural proteins and focus more on custom-designed proteins at the atomic level.
Contacts: Enrique Marcos and Benjamin Basanta
Research conducted by: E. Marcos (Univ. of Washington and Barcelona Institute of Science and Technology); B. Basanta, T.M. Chidyausiku, D.-A. Silva, and J. Dou (Univ. of Washington); Y. Tang, G. Liu, G.V.T. Swapna, R. Guan, R. Xiao, and G.T. Montelione (Rutgers Univ. and Northeast Structural Genomics Consortium); G. Oberdorfer (Univ. of Washington and Univ. of Graz, Austria); J.H. Pereira, B. Sankaran, and P.H. Zwart (Berkeley Lab and Joint BioEnergy Institute); and D. Baker (Univ. of Washington and Howard Hughes Medical Instiute).
Research funding: Howard Hughes Medical Institute, Defense Threat Reduction Agency, National Institutes of Health, European Commission (via Marie Curie International Outgoing Fellowships), Pew Charitable Trusts, Mexican National Council of Science and Technology (CONACYT), and U.S. Department of Energy, Office of Science, Basic Energy Sciences program (DOE BES). Operation of the ALS is supported by DOE BES.
Publication about this research: E. Marcos, B. Basanta, T.M. Chidyausiku, Y. Tang, G. Oberdorfer, G. Liu, G.V.T. Swapna, R. Guan, D.-A. Silva, J. Dou, J.H. Pereira, R. Xiao, B. Sankaran, P.H. Zwart, G.T. Montelione, and D. Baker, “Principles for designing proteins with cavities formed by curved β sheets,” Science 355, 201 (2017). doi:10.1126/science.aah7389
ALS SCIENCE HIGHLIGHT #350
![]()
![]()