Retinitis pigmentosa (RP) is a group of genetic disorders that affect the retina’s ability to respond to light. This inherited disease causes a slow loss of vision, beginning with decreased night vision and loss of peripheral side vision. One of the causes of this disease is the misfolding and aggregation of a protein called opsin found in the retina of the eye.
Using Fourier-transform infrared (FTIR) microspectroscopy at Beamline U2B at NSLS (Brookhaven) and at Beamline 1.4 at the ALS (Berkeley), scientists studied two mutants of opsin that misfold and aggregate.
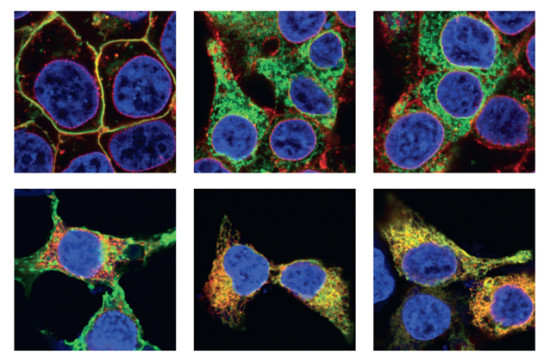
The samples were a cell culture model of RP, in which cells from the HEK293 cell line were cultured in the lab and transfected with DNA coding for mutant opsins. This cell culture model expressed the mutant proteins fused to a fluorescent protein called YFP. The fused fluorescent protein allowed the scientists to use a fluorescence microscope to precisely identify the location of the protein in the cell.
The aggregated opsin mutants are toxic and cause the photoreceptor cells to die, causing retinal degeneration. A better understanding of the structures formed by these mutants is necessary to be able to develop targeted therapeutics to combat the formation of these aggregates. This study provides a first glimpse into the structural changes that result from point mutations in opsin that cause misfolding and aggregation.
Work performed at Beamline U2B at NSLS and at Beamline 1.4 at ALS. The ALS is happy to support the IR community from NSLS as part of the transition program to continue their research while the infrared beamlines at NSLS-II are constructed.
Lisa M. Miller, Megan Gragg, Tae Gyun Kim, and Paul S.-H. Park, “Misfolded opsin mutants display elevated β-sheet structure,” FEBS Letters 589, 3119 (2015).
![]()